
Keywords: quality standards, transfection reagents, cGMP transfection reagent, cGMP manufacturing, viral vectors, raw materials, batch record,
Viral vectors have emerged as the most efficient gene delivery systems to treat a variety of life-threatening diseases, with the number of new viral vectors being tested in clinical trials growing exponentially. As patient safety comes first, their quality and safety are paramount whether they are used directly (in vivo) or indirectly (ex vivo) to deliver therapeutic genes into the human body. To ensure viral vectors used in clinical trials are fit for purpose and comply with quality standards and regulatory requirements, they should be produced under current Good Manufacturing Practices (cGMP), a quality system that certifies pharmaceuticals drug is adequately manufactured, tested and dosed to ensure optimal safety and effectiveness.
cGMP in conjunction with GDP (good documentation practices) cover the entire manufacturing process, from facilities & equipment to raw materials and personnel training. They also cover the quality controls for production, processing, packaging and storage of a drug. Regulatory agencies dealing with drugs and finished pharmaceuticals, such as US Food & Drugs Administration (FDA) or the European Medicines Agency (EMA), are responsible for protecting and promoting patient safety through regulation and supervision during the whole process of developing, testing, manufacturing, holding of and commercialisation of drugs. The regulations provide practical guidance on how to control manufacturing, testing, and quality assurance of pharmaceutical drugs, to ensure that they contain the ingredients and have the strength they claim to have to be safe for human use. cGMP compliance also ensures the drug’s critical attributes (namely safety, identity, quality, purity and potency) are met during manufacturing.
While the critical attributes of the final product are needed for compliance, the facilities themselves and the documentation also need to fulfil certain criteria:
- Cleanliness – The manufacturing must be done in clean and hygienic areas, and these should be monitored regularly.
- Environmental controls- The environmental conditions must be such that cross-contamination is prevented.
- Trained personnel – Operators must be qualified and trained to perform the tasks required.
- Process control – All processes must be clearly defined and controlled.
- Documentation (Batch record) – Processes must follow the Good Documentation Practices (GDP).
- Full traceability – There should be data history regarding raw and starting materials, the manufacturing process including the validation of manufacturing steps and analytical methods for quality control (QC) as well as the use and validation of equipment, distribution, serialisation, and the track & trace support.
- Data integrity – The data should be complete, consistent, and accurate. It should be attributable, legible, contemporaneously recorded, original or true copy and accurate (ALCOA). Data integrity also included in the creation, modification, processing, maintenance, archival, retrieval, transmission, and disposition of data after the record’s retention period ends.
The importance of using cGMP grade raw materials to minimize risk
The demand for viral vector manufacturing is increasing exponentially due to the success of in vivo and ex vivo gene therapies in Phase I/II & III clinical trials and the increasing number of novel therapies under development. Nevertheless, manufacturing of viral vector at scale presents two main challenges that could jeopardize compliance with cGMP regulations: (i) the sourcing of raw materials and (ii) the management of the supply chain. Having unlimited access to high quality raw and starting materials, such as transfection reagent, cell lines, media components, growth factors, serum, consumables and excipients is crucial. Therefore, it is extremely important to confirm the identity, purity, and quality of raw materials prior to starting the viral vector manufacturing, to guarantee the efficacy of viral vectors and to ensure batch to batch reproducibility. This will also ensure patient safety is considered and safeguarded from the start of the manufacturing process.
Raw materials for viral vector manufacturing need to be of the highest quality as they will be scrutinized thoroughly. They must be accompanied by a Certificate of Analysis, a Certificate of Origin to confirm identity and purity, and a certificate of compliance. This applies to cell culture media and other compounds as well as to plasmid DNA and transfection reagents, both of which also need to be high quality. Producing such cGMP grade materials require expertise and thus, there is only a limited number of suppliers that produce cGMP-grade plasmid DNA and even fewer that produce transfection reagents at cGMP grade. Viral vector manufacturers will need to evaluate and qualify those suppliers carefully, to ensure that their final product complies with quality standards and regulatory requirements. To do so, manufacturers usually employ a risk-based approach and methodology for manufacturing (QbD or Quality by Design). As part of this approach, they evaluate the availability and supply of raw and starting materials by mapping what materials become critical at which step of the manufacturing process to mitigate the risk of manufacturing failure.
In addition to using high quality raw materials, viral vector manufacturers must ensure the critical attributes of the final product with patient safety in mind. Failure to produce high quality viral vectors in a timely fashion may put patients’ lives at risk if they do not receive the life-saving treatments they need in time. For ex vivo therapies, any delays with cell engineering might burden an already lengthy and often time-critical cell therapy product manufacturing process. So, whether the viral vectors are delivered directly into patients or used to modify patients’ cells ex vivo, the viral vector batch must be produced to the highest quality for patient safety.
Viral vector manufacturers and developers of new therapeutics need to mitigate other additional risks. For example, lack of traceability for raw materials, non-controlled or un-notified manufacturing changes or failed batches significantly impacts the timelines for lengthy regulatory approvals and delays clinical advancement. This in turns increases the risk of financial loss which may further delay clinical trial progress, and even hinder subsequent rounds of funding. Thus, similarly to other risks, financial loss is best mitigated from the start, by for example ensuring the quality and cGMP-grade of raw materials.
Viral vector manufacturing steps
The production of viral vectors involves several steps including upstream, downstream processes and aseptic filling of the final product (Figure 1). The major bottlenecks during viral vector manufacturing are the upstream productivity and downstream vector yield, both of which will significantly impact the final viral vector titer.
Transfection (plasmid DNA delivery into the cells with the aid of a transfection reagent) is a key step ensure upstream productivity: the efficiency of the transfection step is critical to produce high amounts of viral vectors at scale and to a degree of reproducibility that will satisfy clinical demand, quality, and regulatory requirements. Therefore, the risk associated with transfection in this process is very high and failing to source any of the components at high quality and in large quantities may have a detrimental impact on the production of therapeutic viral vectors. As a result, it is extremely important to select a reliable supplier for cGMP material and to use cGMP grade plasmid DNA and transfection reagents early in the development of a larger scale manufacturing process. It may also be advantageous to choose a supplier that provides both non-GMP and cGMP grade transfection reagent for large scale viral vector manufacturing. Those suppliers provide expert support and robust scale-up protocols which facilitate process upscale, preventing delays in manufacturing timelines.
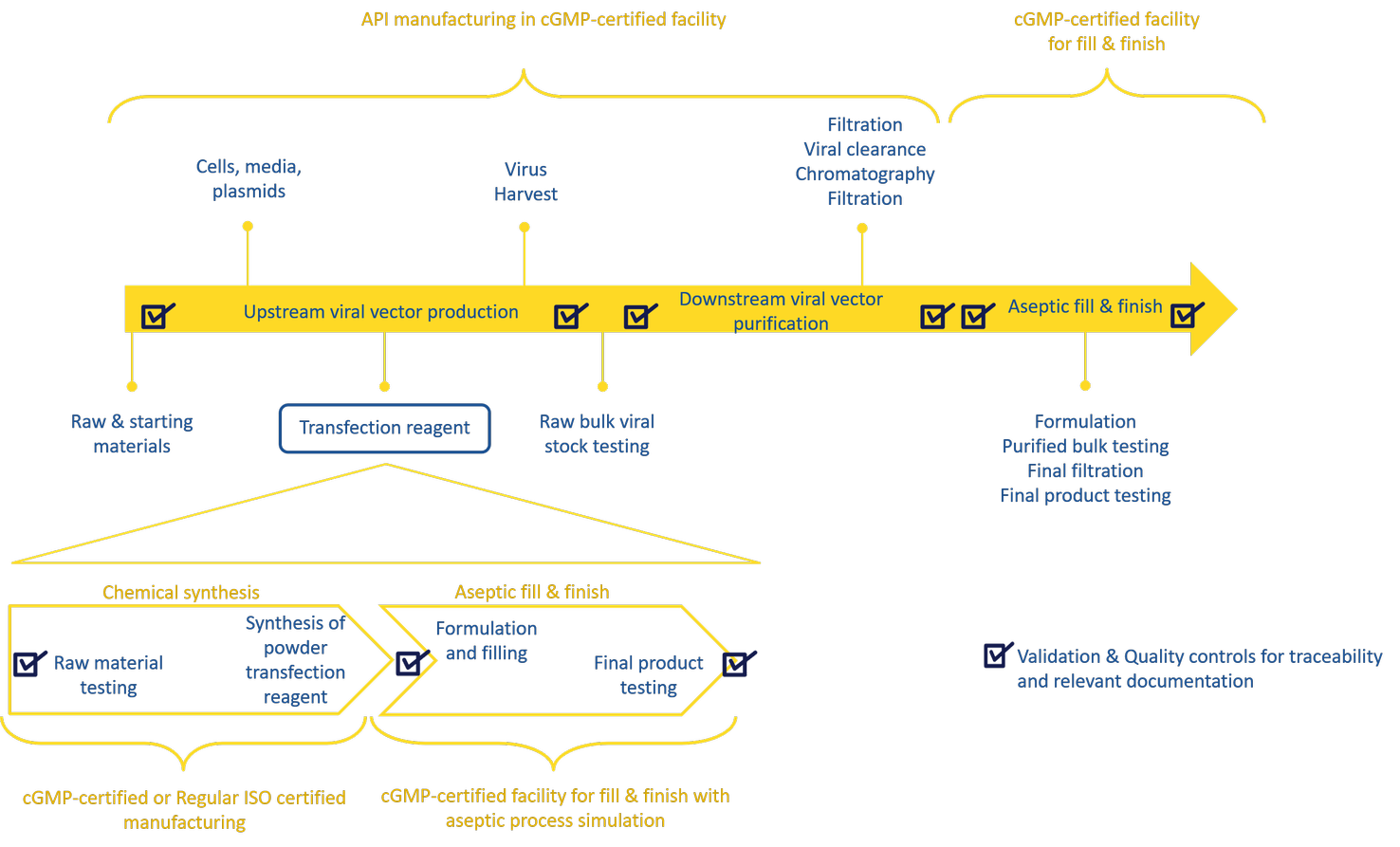
Figure 1: Gene therapy vector manufacturing process workflow:
The production of viral vectors involves several steps: upstream expression, downstream purification and formulation followed by fill & finish steps. The overview of the manufacturing steps of a cGMP transfection reagent are indicated in the bottom half.
Scrutinize how transfection reagents are manufactured: what does GMP grade really mean?
Manufacturing of transfection reagents follows a series of steps from chemical synthesis to formulation and fill & finish of the final product (Figure 1). Suppliers usually manufacture non-GMP transfection reagents in cleanrooms. For cGMP compliant material, some suppliers, such as Polyplus-transfection, produce fully traceable cGMP grade transfection reagents for viral vector manufacturing in compliance with guidelines from start to finish; while others only partially manufacture to cGMP standards, performing a shortcut whereby the synthesis is performed in a cleanroom and only the fill & finish in carried out in a GMP facility. The final product is still cGMP, but the manufacturing process is not in its entirety.
cGMP grade transfection reagents should be produced and formulated following strict guidelines, namely, Eudralex volume 4 Part II in Europe and CFR Part 210 in the United States. Following synthesis, they require extensive characterization and quality control testing ensuring batch-to-batch consistency, traceability and product specifications (identity, purity, potency, safety, and quality). The fill & finish steps for transfection reagents are conducted following Annex 1 EU cGMP manufacturing guidelines which relates to manufacturing of sterile products and includes time consuming aseptic process simulations, a legal requirement if you comply with Annex 1. Consistent with cGMP regulations, with every single batch of cGMP grade transfection reagent, a so called “full documentation” is generated, which details how the batch was produced, including information relating to starting material, equipment, training of personnel, process, cleaning and of course the quality controls related to the batch. This full documentation batch record supports the batch release by the Quality Assurance department guarantying the batch compliance. The resulting product can then be used as a qualified raw material in a cGMP process such as viral vector manufacturing for clinical use.
So, what are the implications of using a transfection reagent synthesized in a standard (non-GMP) facility but for which the fill & finish steps are completed under cGMP compliance? The limitation of using such reagents includes the lack of full traceability of the manufacturing process and the incomplete batch record in conjunction with the financial and regulatory risk for both the manufacturer and the developer, in case of a failed batch. In addition, the early adoption of cGMP grade reagents in a manufacturing process leads to significant time saving during therapeutic viral vector development. In summary, the advantages of using cGMP grade transfection reagents from the beginning of process development are three-fold:
- By working with suppliers of cGMP reagents at the preclinical stage, the transition from laboratory scale to full manufacturing scale is likely to be facilitated, with robust and easily translatable protocols. This will accelerate process development for viral vector manufacturing, reducing both time and costs associated with optimisation.
- The use of a qualified raw material for transfection greatly improves the reproducibility of the transfection step and facilitates batch to batch consistency impacting viral vector yields and timelines, critical bottlenecks during large-scale viral vector manufacturing.
- Full traceability of the entire manufacturing process is an invaluable advantage. The importance of full traceability of the transfection reagent as a qualified raw material cannot be stressed enough, as it is a crucial risk mitigation strategy in the risk-based approach followed during the manufacturing of viral vectors for clinical use.
In conclusion, the importance of cGMP-grade raw materials in the manufacturing of viral vectors should not be overlooked. It is particularly critical in the regulatory context and for cGMP compliance to follow the best risk mitigation strategies to ensure patient safety; such a strategy relies on following best manufacturing practises and using fully traceable raw materials to avoid batch failure. Therefore, developers of in vivo and ex vivo gene therapies should evaluate the quality of cGMP grade raw and starting materials for the manufacturing of their therapeutic viral vectors with the highest scrutiny and avoid any compromise to save costs.
(To receive the next articles of our GMP expertise series directly in your mailbox, subscribe to our cGMP VIP list)